Exploring the Thermodynamic Properties of Doxepin Isomers at H1 Receptor Binding




Introduction
The histamine H1 receptor (H1R) plays a critical role in various physiological processes, hence being a prominent target for modern drug development. This study, led by a team from Tokyo University of Science, investigates the interaction between doxepin isomers and H1R, utilizing isothermal titration calorimetry (ITC) and molecular dynamics (MD) simulations to quantitatively analyze their thermodynamic parameters.
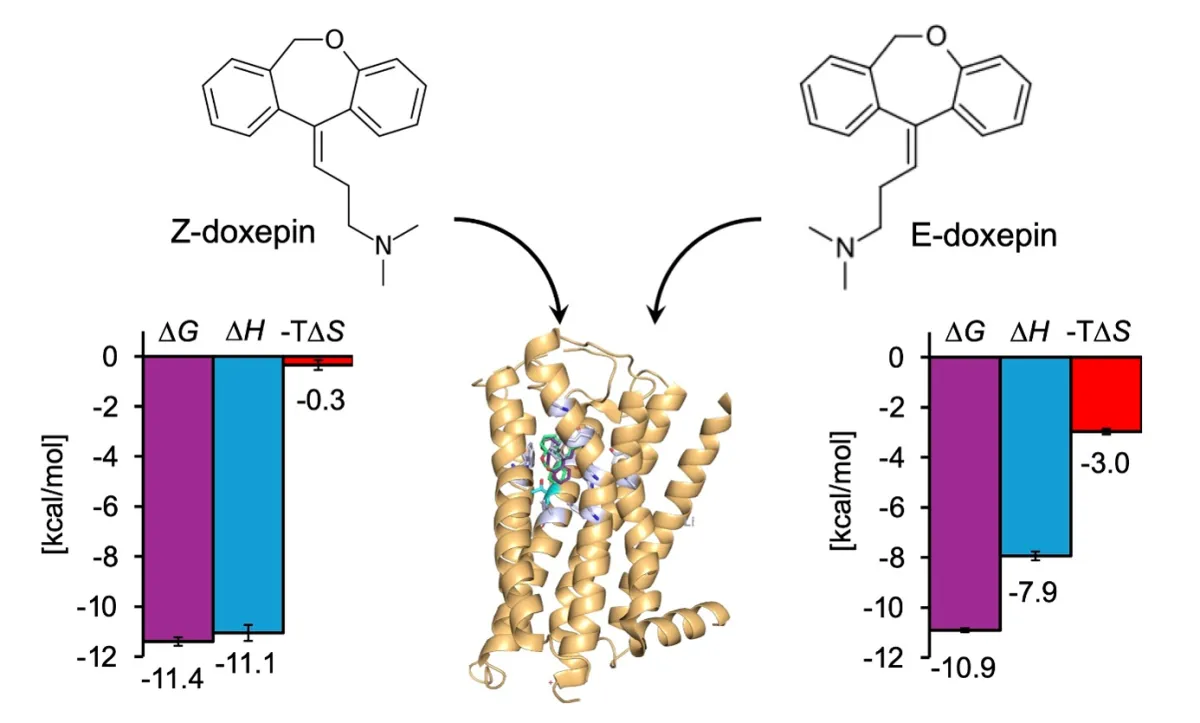
Doxepin, a tricyclic antidepressant, exists in two geometric isomers: E-doxepin and Z-doxepin. Previous findings highlighted that Z-doxepin binds with approximately five times greater affinity to H1R compared to E-doxepin, with a specific residue (Thr1123.37) being pivotal for this selectivity. Hence, this study aims to delve deeper into the enthalpy-entropy compensation dynamics of these isomers to showcase the intricacies of their binding.
Research Overview
Engaging both ITC and MD simulations, the researchers successfully quantified the thermodynamic interactions of H1R with E-doxepin and Z-doxepin. Through this dual approach, the team discovered distinct thermodynamic profiles for both isomers when binding to H1R. In their analysis, the enthalpy-entropy compensation mechanism was revealed as crucial in understanding the binding affinity, particularly how Z-doxepin's spatial configuration significantly restricts its structural diversity compared to E-doxepin, thereby influencing its interaction with the receptor.
G protein-coupled receptors (GPCRs), such as H1R, constitute the largest protein family within the human body and are vital in mediating diverse cellular signaling. Over 30% of available pharmaceuticals target GPCR, highlighting their crucial role in drug discovery. Recent understandings suggest that drug design should leverage not only the binding affinities but also the underlying thermodynamic components of enthalpy and entropy during receptor-ligand interactions.
In the present study, the researchers used a budding yeast expression system to prepare the receptors and measure the direct binding interactions. Notably, they observed that the binding interactions showed no significant differences in affinity between the wild-type and mutated H1R variants; however, stark disparities existed in the contributions from enthalpy and entropy. For wild-type H1R, the binding was primarily enthalpy-driven, governed by non-polar interactions such as van der Waals forces.
On the other hand, in the case of mutated H1R, the enthalpic contributions were minimal, and the entropy became predominant. This suggests that Thr1123.37 is a critical element in stabilizing doxepin interactions with H1R through enthalpic stabilization. When analyzing the thermodynamic parameters for each isomer in the wild-type H1R context, Z-doxepin exhibited a significant enthalpy gain and a corresponding entropy loss compared to E-doxepin.
Molecular Dynamics Analysis
Using MD simulations, thorough cluster analyses of the spatial structures of both isomers before and after receptor binding revealed similar patterns in cluster distribution regarding the wild-type and mutant H1R interactions. However, when bound to wild-type H1R, Z-doxepin's spatial configurations tended to converge into fewer clusters, highlighting its preferred orientation and interaction dynamics with the receptor, driven by Thr1123.37’s hydroxyl group.
This suggests a mechanism where Z-doxepin not only stabilizes a specific spatial structure upon binding but achieves a more substantial enthalpic benefit in exchange for a reduction in entropy. Such findings align with previous results from ITC experiments, emphasizing that Z-doxepin's interaction with wild-type H1R yields higher enthalpic gains and a more considerable entropy loss compared to E-doxepin.
Implications for Drug Design
Professor Mitsunori Shiroishi, who led the study, articulated that the metrics derived from the enthalpy-entropy compensation mechanism will serve as new guidance in GPCR-targeted drug discovery. The meticulous analysis of molecular interactions and dynamic simulations promises advancements in developing drugs that exploit stereochemical characteristics and cater to mutations in GPCRs, aligning with the growing demand for precision medicine.
Furthermore, this strategic investigation method also positions itself as a reference for optimizing lead compounds and integrating artificial intelligence into drug development pipelines. The outcomes of this research underline the necessity for thoughtful consideration of structural attributes when designing ligands to enhance therapeutic profiles.
Conclusion
This study not only advances the foundational understanding of doxepin's interaction with H1R but also sets critical precedents for future exploration within GPCR pharmacology. As pharmaceutical sciences progress, the implications of such research will undoubtedly reverberate throughout the industry, heralding a new era of targeted therapy and individualized treatment strategies.
Topics Health)

【About Using Articles】
You can freely use the title and article content by linking to the page where the article is posted.
※ Images cannot be used.
【About Links】
Links are free to use.