Streamlining Protein Function Prediction Using Molecular Simulation and Language Models




Streamlining Protein Function Prediction Using Advanced Techniques
In a groundbreaking approach, researchers from the National Institute of Advanced Industrial Science and Technology (AIST) have developed a novel method to predict the functional values of proteins with high accuracy using limited experimental data. By combining molecular simulations with protein language models, this new technique represents a significant advancement in the field of artificial intelligence and molecular biology.
The Significance of Protein Function Prediction
Proteins play a vital role in various biological processes, serving as enzymes, antibodies, and structural components in living organisms. Their ability to facilitate chemical reactions, recognize specific molecules, and perform numerous other functions makes them essential in both industrial and medical applications. The conventional methods for predicting protein functions typically rely on extensive datasets garnered from experimental results, which can be both time-consuming and resource-intensive.
Although machine learning methodologies have progressively been adopted to predict protein functions, they often require significant amounts of experimental data to train models effectively. This necessity poses a challenge during scenarios where only a few data points are available.
Innovative Methodology Development
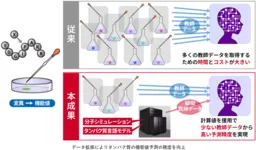
The researchers, led by Teppei Deguchi, have introduced an innovative approach that leverages computational simulations alongside artificial intelligence to expand training datasets. This dual strategy utilizes calculated functional values as pseudo-teacher data to complement minimal experimental datasets. Previously, this approach focused solely on predicting protein stability, but the researchers have broadened its applications to include crucial properties such as binding affinity, enzyme activity, and toxicity.
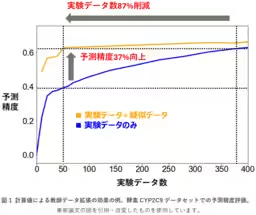
This groundbreaking study has been noted for demonstrating that, even when only 50 experimental data points are available, the inclusion of approximately 4,600 additional simulated data points can boost predictive accuracy by 37%. Such findings highlight the potential for enhancing computational efficiency and reducing dependency on large datasets.
Background and Rationale
Proteins are made up of amino acids linked together in unique sequences, and by modifying these sequences, scientists can create functional proteins with improved stability and activity. To design proteins that meet specific functional criteria efficiently, there has been a push towards incorporating machine learning methodologies to predict protein functions accurately.
Historically, protein engineers relied on experimental data pairs, consisting of amino acid sequences and their functional assessments, to train machine learning models. Due to the time and resources required for experiments, obtaining comprehensive training datasets often proves challenging, making the exploration of alternative approaches to expand these datasets essential.
Using molecular simulations allows for the prediction of a protein's stability by computing its properties and behaviors on a computer. Additionally, protein language models, which treat amino acid sequences like natural language, can be trained on vast databases to estimate protein functionalities without relying solely on experimental input.
Combining Techniques for Enhanced Results
The successful integration of both molecular simulations and protein language models aims to enhance data-driven predictions of protein functionalities. In this study, the researchers aimed explicitly to improve predictive accuracy despite the constraints of limited experimental inputs.
By creating pseudo-teacher datasets with hundreds or thousands of simulated sequences, their methodology allows for a more robust training process. This innovative combination not only captures the physical principles governing molecular interactions but also utilizes AI capabilities to interpret variations in amino acid sequences systematically.
The researchers also designed algorithms to automatically adjust the trustworthiness of these pseudo-teacher datasets, developing judgment algorithms to minimize risk during instances where predictive accuracy might decrease.
Real-World Applications and Future Directions
Looking ahead, the researchers are focused on translating their method into practical applications for antibody and enzyme development. Furthermore, they aspire to deploy their technology into a functional protein design system that can be utilized in university and corporate laboratories.
This advancement could significantly revolutionize the landscape of functional protein development and enhance the efficiency of bioengineering.
Conclusion
The findings of this research, which illustrate the synergistic potential of molecular simulation and protein language models, offer promising pathways for high-accuracy protein function prediction. As the demand for efficient protein design rises in various sectors, the significance of this research becomes increasingly pronounced, heralding a new horizon in protein engineering.
For more details, refer to the publication in Briefings in Bioinformatics on October 10, 2025:
Title: Data-efficient protein mutational effect prediction with weak supervision by molecular simulation and protein language models.
Authors: Teppei Deguchi, Nur Syatila Ab Ghani, Yoichi Kurumida, Shinji Iida, Kaito Kobayashi, Yutaka Saito.
Press Release Link
Topics Consumer Technology)
【About Using Articles】
You can freely use the title and article content by linking to the page where the article is posted.
※ Images cannot be used.
【About Links】
Links are free to use.