Understanding the Mechanism for Cellular Resistance to Alovudine: Implications for Cancer Treatment




Unveiling Cellular Resistance Mechanisms to Alovudine
Researchers from Tokyo Metropolitan University have made significant strides in understanding how human cells resist the toxicity of alovudine, a nucleoside analog previously investigated for treating HIV. Despite its potential, alovudine's high cell toxicity led to the discontinuation of its clinical trials. However, it is now used in PET scans for detecting tumors using its fluorine-18 labeled compound as a tracer.
Alovudine mimics thymidine in structure and can be incorporated into viral genomes during replication, thus halting viral proliferation. Until recently, the mechanisms behind how human cells counteract its toxicity remained poorly understood. In a groundbreaking study led by Professor Kouji Hirota and his team, they discovered that a DNA repair enzyme known as flap endonuclease 1 (FEN1) plays a vital role in reducing alovudine's toxicity by removing the drug from the replicating DNA strand.
Research Findings
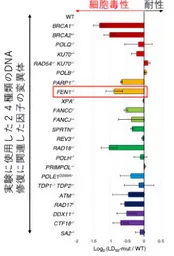
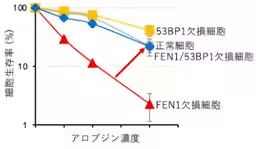
The research team initially set out to investigate the toxicity of alovudine across various cellular mutations linked to DNA repair. Utilizing 24 mutant genes related to DNA repair, they pinpointed FEN1 as a critical player in cellular resistance. Cells deficient in the FEN1 gene exhibited significantly heightened toxicity when treated with alovudine, indicating that FEN1 functions as a protective mechanism against the drug's harmful effects.
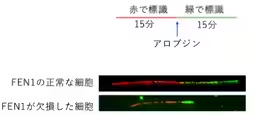
Moreover, the researchers observed that the replication speed of DNA was notably slower in FEN1-deficient cells following alovudine treatment, signifying that the lack of FEN1 impairs the normal DNA replication process under stress from nucleoside analogs.
An interesting finding revealed that the increased sensitivity of FEN1-deficient cells could be mitigated by simultaneous loss of a gene coding for another DNA repair protein, 53BP1. When both FEN1 and 53BP1 were absent, the protective effects of FEN1 on cells treated with alovudine were largely restored, indicating a complex interplay between these proteins in managing toxicity.
In normal cells, FEN1's activity helps eliminate abnormal DNA induced by alovudine. If this removal process falters, the accumulated drug leads to an abnormal buildup of 53BP1 within the genome, eventually resulting in severe DNA damage and cell death.
Implications for Cancer Treatment
The findings from this study suggest a promising link between FEN1's function and the treatment of various cancers, particularly those with BRCA gene mutations associated with breast cancer. Since FEN1 mutations are prevalent in several cancer types, understanding its role provides a potential biomarker for identifying patients who might benefit from alovudine therapy. The discovery that enhancing FEN1's activity might improve alovudine's therapeutic effects presents a novel direction for cancer treatments.
Furthermore, this research paves the way for developing new anti-cancer drugs targeting FEN1's nuclease activity. Treatments that augment alovudine's efficiency in killing cancer cells harboring FEN1 mutations could lead us towards more effective combating strategies against aggressive tumors.
The implications of this discovery are vast, from clinical diagnostics recognizing patients for targeted treatments to advancing novel anti-cancer drug development strategies. As research into the synergy between FEN1 and alovudine progresses, a clearer picture of their interaction may inspire foundational changes in cancer therapy.
Conclusion
Professor Hirota's team has opened a new frontier in cancer research by elucidating the cellular defenses against a significant therapeutic agent, alovudine. Their findings are expected to have far-reaching effects on the future of cancer treatment, highlighting the importance of understanding cellular mechanisms to enhance therapeutic effectiveness. Future studies should focus on investigating the effects of alovudine on various cancer types, including solid tumors, and paving the path from theory to practice, directly benefiting patient care.

Topics Health)

【About Using Articles】
You can freely use the title and article content by linking to the page where the article is posted.
※ Images cannot be used.
【About Links】
Links are free to use.